Description
Copyright infringement not intended
Picture Courtesy: lsdssindia.org
Context: Pompe disease is a rare genetic disorder that affects the muscles, causing progressive weakness and impacting various bodily functions.
About Pompe disease
- Pompe disease is a complex condition stemming from a genetic defect affecting the breakdown of glycogen in cells.
Genetic Basis and Enzyme Dysfunction
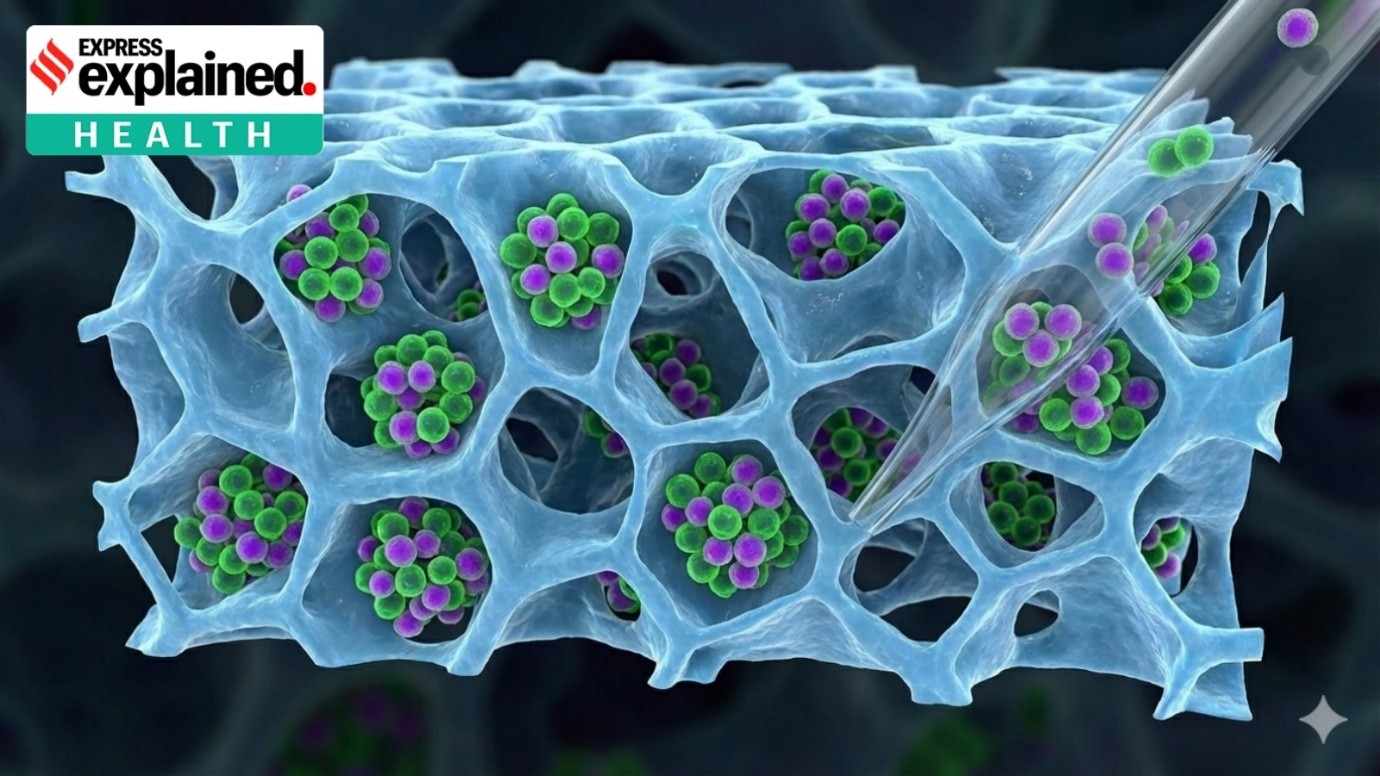
- Pompe disease arises due to mutations in the GAA gene, leading to deficient or dysfunctional acid alpha-glucosidase (GAA) enzymes. GAA normally resides within lysosomes, cellular compartments responsible for breaking down complex molecules like glycogen into simpler forms for energy usage.
- When GAA malfunctions or is absent, glycogen accumulates within these lysosomes, causing cell damage, especially impacting the muscles and heart.
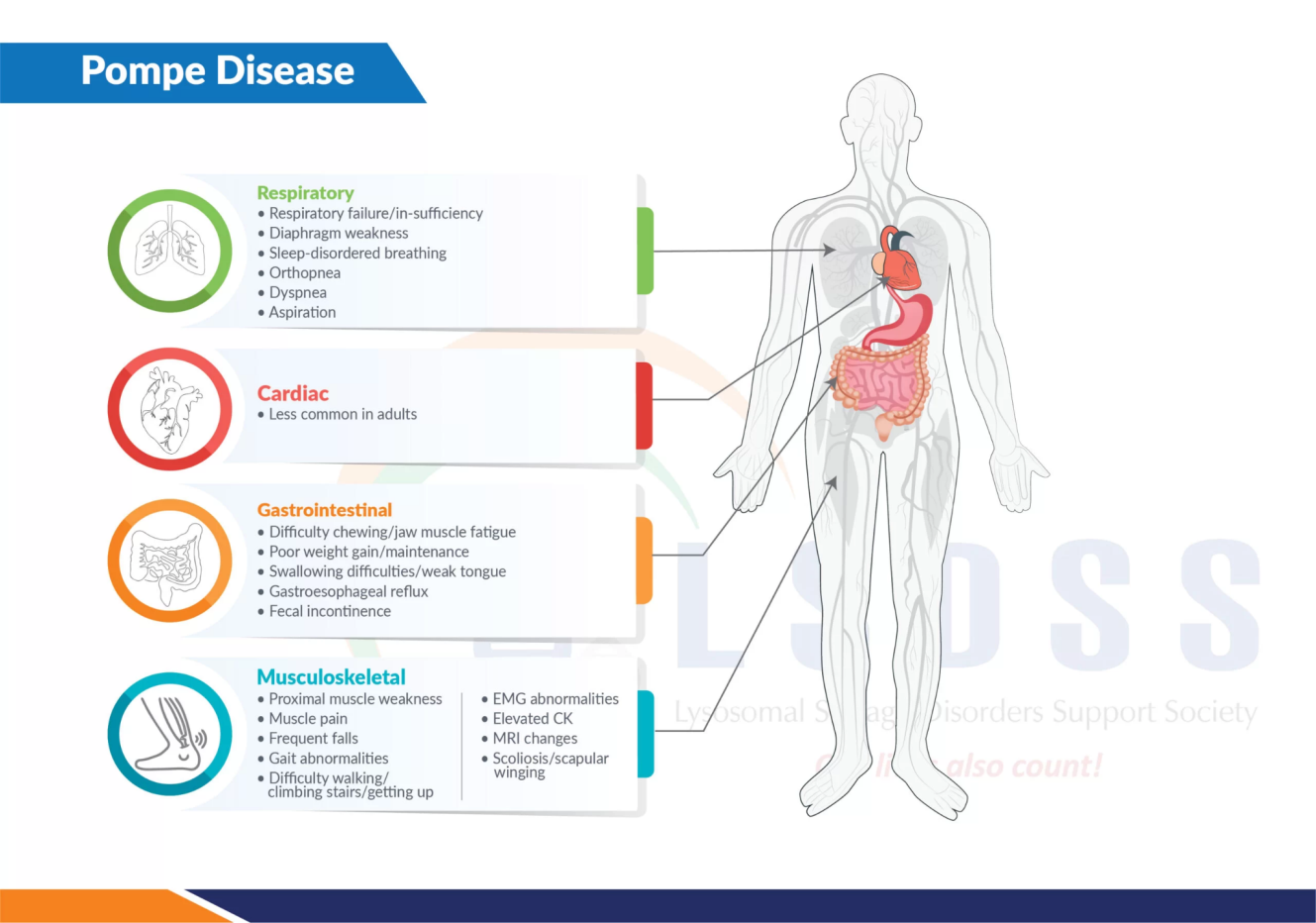
Clinical Presentation and Variants
Infantile-Onset Pompe Disease
- Typically manifest in infancy with severe cardiac and musculoskeletal complications.
- Rapidly progressive muscle weakness, enlarged heart (cardiomyopathy), respiratory difficulties, feeding challenges, and failure to thrive.
- Without treatment, infantile-onset Pompe disease can be fatal before the age of one.
Late-Onset Pompe Disease
- Can occur from childhood to adulthood with varying symptoms and progression.
- Primarily affects skeletal muscles leading to progressive weakness, impacting mobility, breathing, and swallowing.
- Some cases may include mild cardiac issues or cognitive impairment.

Diagnostic Approach
- Identifying characteristic signs and symptoms.
- Measuring GAA activity in the blood and genetic testing to confirm mutations in the GAA gene.
- Sometimes conducted to observe glycogen accumulation within muscle cells.
Treatment Options
- Enzyme Replacement Therapy (ERT): The primary treatment involves regular infusions of synthetic GAA to reduce glycogen buildup and mitigate symptoms. However, it doesn’t reverse existing damage.
- Supportive Therapies: Physical therapy, respiratory support, nutritional assistance, orthopaedic interventions, and surgical procedures aim to manage symptoms and enhance the quality of life.
- Other Supportive Measures: Genetic counselling, psychological support, and community engagement through patient organizations can provide invaluable support.
Challenges and Ongoing Research
- While ERT can improve outcomes, it's not a cure and might present side effects.
- Continual exploration of new treatment avenues and preventive measures to enhance the management and outlook for Pompe disease.

Conclusion
- Pompe disease is a complex disorder with severe implications, necessitating comprehensive care, early diagnosis, and ongoing management. While treatment options exist, ongoing research aims to expand therapeutic possibilities and improve the overall prognosis for affected individuals.
|
PRACTICE QUESTION
Q. Case Study:
Emma, a 4-month-old infant, presents with poor muscle tone, difficulty feeding, and respiratory problems. After several tests, the doctors diagnosed her with Pompe disease. Which of the following best describes the underlying cause of Pompe disease?
A) Deficiency of alpha-glucosidase enzyme
B) Mutation in the dystrophin gene
C) Absence of hexosaminidase A enzyme
D) Mutation in the CFTR gene
Correct Answer: A
Explanation:
Pompe disease, or glycogen storage disease type II, arises from alpha-glucosidase enzyme deficiency, impeding glycogen breakdown in cell lysosomes. The enzyme's role in converting glycogen to glucose is pivotal for waste disposal. Insufficient alpha-glucosidase causes glycogen buildup within lysosomes, predominantly impacting muscle cells. Symptoms manifest early in infancy, exerting effects across bodily systems, notably on skeletal and cardiac muscles due to glycogen accumulation. Progressive muscle weakening ensues, accompanied by severe respiratory complications, posing life-threatening risks.
|